I’m reaching out to discuss considerations regarding logbook usage.
I’d like to inquire if there are specific guidelines for employing logbooks in the pharmaceutical industry.
For every batch released for compression, we conduct physical parameter tests, including uniformity of weight, average weight, DT, hardness, friability, etc. Subsequently, we record this data in dedicated logbooks. Additionally, after each In-Process Control (IPC), which involves further physical parameter testing, we meticulously document all IPC data. Is this documentation truly necessary?
Furthermore, I’m seeking guidance on the frequency of IPC testing. Is it acceptable to permit batch compression solely based on tests conducted by production personnel, without a preliminary assessment by IPC personnel? Your insights into industry practices or guidelines would be highly appreciated.
Here are the answers & clarifications to your questions.
Question:
I’m reaching out to discuss considerations regarding logbook usage.
I’d like to inquire if there are specific guidelines for employing logbooks in the pharmaceutical industry.
Answer:
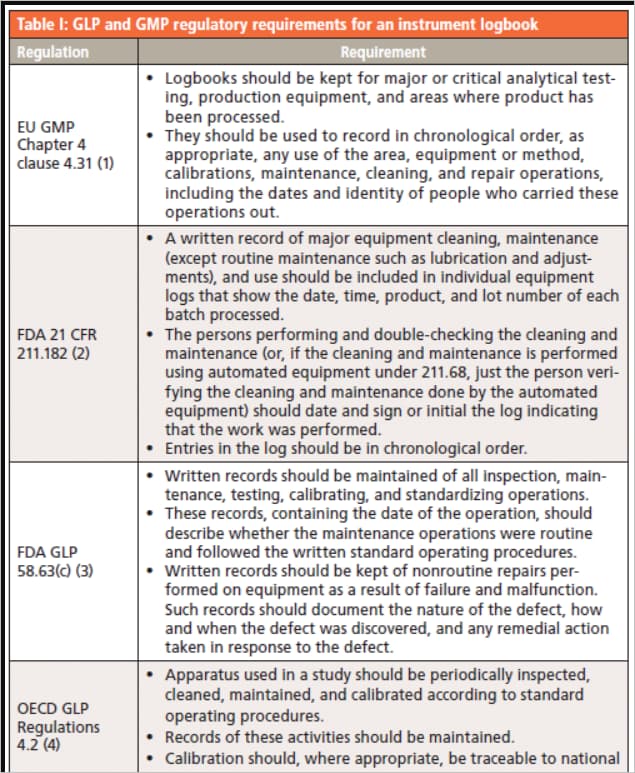
Please find below regulatory references about the requirement for logbooks in the pharmaceutical industry.
Question:
For every batch released for compression, we conduct physical parameter tests, including uniformity of weight, average weight, DT, hardness, friability, etc. Subsequently, we record this data in dedicated logbooks. Additionally, after each In-Process Control (IPC), which involves further physical parameter testing, we meticulously document all IPC data. Is this documentation truly necessary?
Answer:
As per my understanding and the industry practices logbook is not necessary to record the analytical results including those of IPC checks. The analytical results should be entered in the analytical reports and the data should be directly entered in the analyst’s notebook or raw data forms which are controlled documents. The analytical instruments used for testing / analyzing the samples of compressed tablets and other samples should be entered in the individual usage logbooks for analytical instruments maintained in the QC / IPC laboratory.
Question:
Furthermore, I’m seeking guidance on the frequency of IPC testing. Is it acceptable to permit batch compression solely based on tests conducted by production personnel, without a preliminary assessment by IPC personnel? Your insights into industry practices or guidelines would be highly appreciated.
Answer:
It is not acceptable to solely rely on the tests performed by production personnel during the compression of tablets (or any stage of processing of the products). There should be IPC tests performed independently by either QC/IPQC/QA/IPQA as per the internal Quality system of the organization.
Valid in-process specifications and testing frequencies for such characteristics shall be consistent with drug product final specifications and shall be derived from previous acceptable process averages and process variability estimates where possible and determined by the application of suitable statistical procedures where appropriate (as established during process optimization and validation studies). Examination and testing of samples shall assure that the drug product and in-process material conform to specifications.
In-process materials shall be tested for identity, strength, quality, and purity as appropriate, and approved or rejected by the quality control unit, during the production process, e.g., at commencement or completion of significant phases or after storage for long periods.
(Reference: cGMP requirements as per USFDA, 21 CFR part 211.110, Subpart F - Production and Process Controls).
Before compression operation, tablet compression machine setting checks are performed by production personnel and verified or rechecked by IPQC/IPQA persons. Subsequently, the IPQC checks are performed at the defined frequencies by production and QA persons independently as per in-house SOPs. The IPQC results are well documented in the prescribed formats and Batch Manufacturing and packing records.
Thank you. Its very important issue.
But I’m not fully agree with clarification that IPC testing by production personals is not enough. Its depend on specification requirements. If some tests must be performed in production as pH, Humidity ( Mettler) for granulate, hardness, thickness, diameter, weight and disintegration for tablets, and documented, not any additional tests required by QA/QC. Additional tests may be only if some analytical tests are included in spec. ( Bulk density, viscous etc ). Also I don’t see the requirements to add any IPC tests to Release specification- this is only transfer data from IPC to Release.
Hello Alex,
I refer to your comments on my previous communication on the 'In-process checks".
In fact, as per cGMP requirements specified in the USFDA, 21 CFR part 211.110, Subpart F - Production and Process Controls, in-process materials shall be tested for identity, strength, quality, and purity as appropriate, and approved or rejected by the quality control unit, during the production process, e.g., at commencement or completion of significant phases or after storage for long periods.
pharmaceutical industry, accuracy, compliance, and safety are paramount. Indeed, the use of logbooks is a long-standing practice. They are **written records that document the activities, observations, and results of various processes and procedures.